CATEGORIES:
BiologyChemistryConstructionCultureEcologyEconomyElectronicsFinanceGeographyHistoryInformaticsLawMathematicsMechanicsMedicineOtherPedagogyPhilosophyPhysicsPolicyPsychologySociologySportTourism
Protooncogenes, Oncogenes, and Oncoproteins
As often happens in science, the discovery of protooncogenes was not straightforward. These cellular genes were first discovered in their mutated or "oncogenic" forms as "passengers" within the genome of acute transforming retroviruses by the 1989 Nobel laureates Harold Varmus and Michael Bishop. These retroviruses cause rapid induction of tumors in animals and can also transform animal cells in vitro. Molecular dissection of their genomes revealed the presence of unique transforming sequences (viral oncogenes [v-onc]) not found in the genomes of nontransforming retroviruses. Most surprisingly, molecular hybridization revealed that the v-onc sequences were almost identical to sequences found in normal cellular DNA. From this evolved the concept that during evolution, cellular oncogenes were transduced (captured) by the virus through a chance recombination with the DNA of a (normal) host cell that had been infected by the virus. Because they were discovered initially as viral genes, these protooncogenes were named after their viral homologues. Each v-onc is designated by a three-letter word that relates the oncogene to the virus from which it was isolated. Thus, the v-onc contained in feline sarcoma virus is referred to as v-FES, whereas the oncogene in simian sarcoma virus is called v-SIS. The corresponding protooncogenes are referred to as FES and SIS, dropping the prefix.
The viral oncogenes are not present in several cancer-causing RNA viruses. One such example is a group of so-called slow transforming viruses that cause leukemias in rodents after a long latent period. The mechanism by which they cause neoplastic transformation implicates protooncogenes. Molecular dissection of the cells transformed by these leukemia viruses revealed that the proviral DNA is always integrated (inserted) near a protooncogene. One consequence of proviral insertion near a protooncogene is to induce a structural change in the cellular gene, thus converting it into a cellular oncogene (c-onc, or onc). This mode of protooncogene activation is called insertional mutagenesis. Alternatively, strong retroviral promoters inserted in the vicinity of the protooncogenes lead to dysregulated expression of the cellular gene.
Although the study of transforming animal retroviruses provided the first glimpse of protooncogenes, these investigations did not explain the origin of human tumors, which (with rare exceptions) are not caused by infection with retroviruses. Hence the question was raised: Do nonviral tumors contain oncogenic DNA sequences? The answer was provided by experiments involving DNA-mediated gene transfer (DNA transfection). When DNA extracted from several different human tumors was transfected into mouse fibroblast cell lines in vitro, the recipient cells acquired some properties of neoplastic cells. The conclusion from such experiments was inescapable: DNA of spontaneously arising cancers contains oncogenic sequences, or oncogenes. One of the first oncogenic sequences detected in cancers was a mutated form of the RAS protooncogene. This protooncogene is the forbear of v-oncs contained in Harvey (H) and Kirsten (K) sarcoma viruses.
A large number of protooncogenes have been identified during the past 20 years, most of which do not have a viral counterpart. Protooncogenes have multiple roles, participating in cellular functions related to growth and proliferation. Proteins encoded by protooncogenes may function as growth factor ligands and receptors, signal transducers, transcription factors, and cell-cycle components ( Fig. 7-31 ). Oncoproteins encoded by oncogenes generally serve similar functions as their normal counterparts ( Table 7-8 ). However, because they are constitutively expressed, oncoproteins endow the cell with self-sufficiency in growth.[45]
To summarize, protooncogenes may be converted into cellular oncogenes (c-oncs) that are involved in tumor development. Two questions follow: (1) What are the functions of oncogene products, the oncoproteins? (2) How do the normally "civilized" protooncogenes turn into "enemies within"? These issues are discussed below.
Growth Factors.
Many cancer cells develop growth self-sufficiency by acquiring the ability to synthesize the same growth factors to which they are responsive. The protooncogene SIS, which encodes the chain of platelet-derived growth factor (PDGF), is overproduced in many tumors, especially low-grade astrocytomas and osteosarcomas. Furthermore, it appears that the same tumors also express receptors for PDGF and are hence responsive to autocrine stimulation. Although an autocrine loop is considered to be an important element in the pathogenesis of several tumors, in most instances the growth factor gene itself is not altered or mutated. More commonly, products of other oncogenes such as RAS (that lie along many signal transduction pathways) cause overexpression of growth factor genes, thus forcing the cells to secrete large amounts of growth factors, such as transforming growth factor-(TGF-). This growth factor is related to epidermal growth factor (EGF) and induces proliferation by binding to the EGF receptor. TGF-is often domain alter the substrate specificity of the tyrosine kinase and lead to thyroid and adrenal tumors but no involvement of the parathyroid. Complete loss of RET function results in Hirschsprung disease ( Chapter 17 ), in which there is lack of development of intestinal nerve plexuses. In all these familial conditions, the affected individuals inherit the RET mutation in the germ line. Sporadic medullary carcinomas of the thyroid are associated with somatic rearrangements of the RET gene, generally similar to those found in MEN 2B.[46] [47]
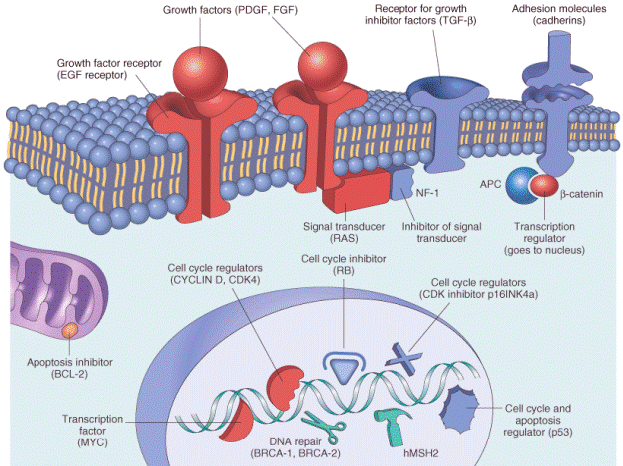
Figure 7-31Subcellular localization and functions of major classes of cancer-associated genes. The protooncogenes are colored red, cancer suppressor genes blue, DNA repair genes
green, and genes that regulate apoptosis purple.
TABLE 7-8-- Selected Oncogenes, Their Mode of Activation, and Associated Human Tumors
| Category | Protooncogene | Mode of Activation | Associated Human Tumor |
| Growth Factors | |||
| PDGF-βchain | SIS | Overexpression | Astrocytome |
| Osteosarcoma | |||
| Fibroblast growth factors | HST-1 | Overexpression | Stomach cancer |
| INT-2 | Amplification | Bladder cancer | |
| Breast cancer | |||
| Melanoma | |||
| TGFα | TGFα | Overexpression | Astrocytomas |
| Hepatocellular carcinomas | |||
| HGF | HGF | Overexpression | Thyroid cancer |
| Growth Factor Receptors | |||
| EGF-receptor family | ERB-B1 (ECFR) | Overexpression | Squamous cell carcinomas of lung, gliomas |
| ERB-B2 | Amplification | Breast and ovarian cancers | |
| CSF-1 receptor | FMS | Point mutation | Leukemia |
| Receptor for neurotrophic factors | RET | Point mutation | Multiple endocrine neoplasia 2A and B, familial medullary thyroid carcinomas |
| PDGF receptor | PDGF-R | Overexpression | Gliomas |
| Receptor for stem cell (steel) factor | KIT | Point mutation | Gastrointestinal stromal tumors and other soft tissue tumors |
| Proteins Involved in Signal Transduction | |||
| GTP-binding | K-RAS | Point mutation | Colon, lung, and pancreatic tumors |
| H-RAS | Point mutation | Bladder and kidney tumors | |
| N-RAS | Point mutation | Melanomas, hematologic malignancies | |
| Nonreceptor tyrosine kinase | ABL | Translocation | Chronic myeloid leukemia |
| Acute lymphoblastic leukemia | |||
| RAS signal transduction | BRAF | Point mutation | Melanomas |
| WNT signal transduction | -catenin | Point mutation | Hepatoblastomas, hepatocellular carcinoma |
| Overexpression | |||
| Nuclear Regulatory Proteins | |||
| Transcriptional activators | C-MYC | Translocation | Burkitt lymphoma |
| N-MYC | Amplification | Neuroblastoma, small cell carcinoma of lung | |
| L-MYC | Amplification | Small cell carcinoma of lung | |
| Cell-Cycle Regulators | |||
| Cyclins | CYCLIN D | Translocation | Mantle cell lymphoma |
| Amplification | Breast and esophageal cancers | ||
| CYCLIN E | Overexpression | Breast cancer | |
| Cyclin-dependent kinase | CDK4 | Amplification or point mutation | Glioblastoma, melanoma, sarcoma |
Oncogenic conversions by mutations and rearrangements have been found in other growth factor receptor genes. Point mutations that activate c-FMS, the gene encoding the colonystimulating factor 1 (CSF-1) receptor, have been detected in myeloid leukemias. In certain chronic myelomonocytic leukemias with the t(12;9) translocation, the entire cytoplasmic domain of the PDGF receptor is fused with a segment of the ETS family transcription factor, resulting in permanent dimerization of the PDGF receptor.
Far more common than mutations of these protooncogenes is overexpression of normal forms of growth factor receptors. In sporadic papillary thyroid carcinomas, c-MET is overexpressed in almost every case.[48] In these tumors, increased expression of c-MET is not caused by gene mutation but results from enhanced transcription of the gene. In some tumors, increased receptor expression results from gene amplification, but in many cases, the molecular basis of increased receptor expression is not fully known. Two members of the EGF receptor family are most commonly involved. The normal form of ERB B1, the EGF receptor gene, usually referred to as EGFR, is overexpressed in up to 80% of squamous cell carcinomas of the lung, in 50% or more of high-grade astrocytomas called glioblastomas ( Chapter 28 ), in 80% to 100% of head and neck tumors, and less commonly, in carcinomas of the urinary bladder and the gastrointestinal tract.[49] [50] In contrast, the ERB B2 gene (also called HER 2/Neu), the second member of the EGF receptor family, is amplified in approximately 25% of breast cancers and in human adenocarcinomas arising within the ovary, lung, stomach, and salivary glands.[51] Because the molecular alteration in ERB B2 is specific for the cancer cells, new therapeutic agents consisting of monoclonal antibodies against ERB B2 have been developed and are currently in use clinically.[49] [51] This type of therapy, directed to a specific alteration in the cancer cell, is called targeted therapy. [52] Another example of very successful targeted cancer therapy is the blockage of receptor tyrosine kinase activity of c-KIT in stromal tumors of the gastrointestinal tract.[53] In these tumors, a mutation in c-KIT, the gene encoding the receptor for stem cell factor (also known as steel factor), constitutively activates the receptor tyrosine kinase, independent of ligand binding.
Date: 2016-04-22; view: 1059
| <== previous page | | | next page ==> |
| Molecular Basis of Cancer | | | Signal-Transducing Proteins. |