CATEGORIES:
BiologyChemistryConstructionCultureEcologyEconomyElectronicsFinanceGeographyHistoryInformaticsLawMathematicsMechanicsMedicineOtherPedagogyPhilosophyPhysicsPolicyPsychologySociologySportTourism
Molecular Basis of Cancer
The literature on the molecular basis of cancer continues to proliferate at such a rapid pace that it is easy to get lost in the growing forest of information. We list some fundamental principles before delving into the details of the molecular basis of cancer.
• Nonlethal genetic damage lies at the heart of carcinogenesis. Such genetic damage (or mutation) may be acquired by the action of environmental agents, such as chemicals, radiation, or viruses, or it may be inherited in the germ line. The term "environmental," used in this context, involves any acquired defect caused by exogenous agents or endogenous products of cell metabolism. Not all mutations, however, are "environmentally" induced. Some may be spontaneous and stochastic.
• A tumor is formed by the clonal expansion of a single precursor cell that has incurred the genetic damage (i.e., tumors are monoclonal). Clonality of tumors can be assessed in women who are heterozygous for polymorphic X-linked markers, such as the enzymes glucose-6-phosphate dehydrogenase (G6PD), iduronate-2-sulfatase and phosphoglycerate kinase. The principle underlying such an analysis is illustrated in Figure 7-26 . The most commonly used method to determine tumor clonality involves the analysis of methylation patterns adjacent to the highly polymorphic locus of the human androgen receptor gene (HUMARA).[33] The frequency of HUMARA polymorphism in the general population is more than 90%, so it is easy to establish clonality by showing that all the cells in a tumor express the same allele. For tumors with a specific translocation, such as in myeloid leukemias, the presence of the translocation can be used to assess clonality. Immunoglobulin receptor and T-cell receptor gene rearrangements serve as markers of clonality in Band T-cell lymphomas, respectively.
• Four classes of normal regulatory genes—the growth-promoting protooncogenes, the growth-inhibiting tumor suppressor genes, genes that regulate programmed cell death (apoptosis), and genes involved in DNA repair—are the principal targets of genetic damage. Mutant alleles of protooncogenes are considered dominant because they transform cells despite the presence of a normal counterpart. In contrast, both normal alleles of the tumor suppressor genes must be damaged for transformation to occur, so this family of genes is sometimes referred to as recessive oncogenes. However, there are exceptions to this rule, and some tumor suppressor genes lose their suppressor activity when a single allele is lost or inactivated.[34] This loss of function of a recessive gene caused by damage of a single allele is called haploinsufficiency. Genes that regulate apoptosis may be dominant, as are protooncogenes, or they may behave as tumor suppressor genes.
• DNA repair genes affect cell proliferation or survival indirectly by influencing the ability of the organism to repair nonlethal damage in other genes, including protooncogenes, tumor suppressor genes, and genes that regulate apoptosis. A disability in the DNA repair genes can predispose to mutations in the genome and hence to neoplastic transformation. Such propensity to mutations is called a mutator phenotype. [35] With some exceptions, both alleles of DNA repair genes must be inactivated to induce such genomic instability; in this sense, DNA repair genes may also be considered as tumor suppressor genes.
• Carcinogenesis is a multistep process at both the phenotypic and the genetic levels. A malignant neoplasm has several phenotypic attributes, such as excessive growth, local invasiveness, and the ability to form distant metastases. These characteristics are acquired in a stepwise fashion, a phenomenon called tumor progression. At the molecular level, progression results from accumulation of genetic lesions that in some instances are favored by defects in DNA repair.

Figure 7-26Diagram depicting the use of X-linked isoenzyme cell markers as evidence of the monoclonality of neoplasms. Because of random X inactivation, all females are mosaics with two cell populations (with G6PD isoenzyme A or B in this case). When neoplasms that arise in women who are heterozygous for X-linked markers are analyzed, they are made up of cells that contain the active maternal (XA ) or the paternal (XB ) X chromosome but not both.
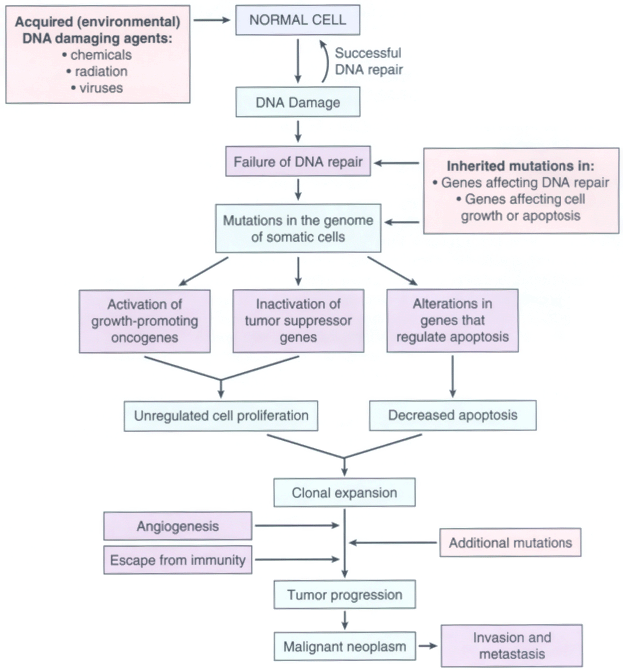
Figure 7-27Flow chart depicting a simplified scheme of the molecular basis of cancer.

Figure 7-28Expression of cyclin-cyclin-dependent kinase (CDK) complexes during the cell cycle. The phases of the cycle are indicated inside the arrows. (Modified from Pollard TD, Earnshaw WC: Cell Biology. Philadelphia, WB Saunders, 2002.)

Figure 7-29Schematic illustration of the role of cyclins, CDKs, and cyclin-dependent kinase inhibitors in regulating the G1 /S cell-cycle transition. External signals activate multiple signal transduction pathways, including those involving the MYC and RAS genes, which lead to synthesis and stabilization of cyclin D (there are several D cyclins, but, for simplification, we refer to them as "cyclin D"). Cyclin D binds to CDK4, forming a complex with enzymatic activity (cyclin D can also bind to CDK6, which appears to have a similar role as CDK4). The cyclin D-CDK4 complex phosphorylates RB, located in the E2F/DP1/RB complex in the nucleus, activating the transcriptional activity of E2F (E2F is a family of transcription factors, which we refer to as "E2F"), which leads to transcription of cyclin E, cyclin A and other proteins needed for the cell to go through the late G1 restriction point. The cell cycle can be blocked by the Cip/Kip inhibitors p21 and p27 (red boxes) and the INK4A/ARF inhibitors p16INK4A and p14ARF (green boxes). Cell-cycle arrest in response to DNA damage and other cellular stresses
is mediated through p53. The levels of p53 are under negative regulation by MDM2, through a feedback loop that is inhibited by p14ARF.

Figure 7-30Mechanism of cell-cycle regulation by RB. In a resting cell, RB is a component of the E2F/DP1/RB complex, which represses gene transcription through the recruitment of histone deacetylase, an enzyme that alters the conformation of chromatin, making it more compact. Phosphorylation of RB by cyclin D-CDK4 removes histone deacetylase from chromatin, allowing the activation of E2F transcriptional activity (RB can also be phosphorylated by cyclin E-CDK2). E2F-mediated transcription of cyclins E and A, and of genes required for DNA replication, permit the passage through the G1 restriction point. (Adapted from Pollard TD, Earnshaw WC: Cell Biology. Philadelphia, WB Saunders, 2002, p. 689.)
TABLE 7-7-- Main Cell-Cycle Components and Their Inhibitors
| Cell-Cycle Component | Main Function |
| Cyclin-Dependent Kinases | |
| • CDK4 | Forms a complex with cyclin D. The complex phosphorylates RB, allowing the cell to progress through the G1 restriction point. |
| • CDK2 | Forms a complex with cyclin E in late G1 , which is involved in the G1 /S transition. Forms a complex with cyclin A at the S phase that facilitates the G2 /M transition. |
| • CDK1 | Forms a complex with cyclin B, which acts on the G2 /M transition. |
| Inhibitors | |
| • Cip/Kip family: p21, p27 | Block the cell cycle by binding to cyclin-CDK complexes. p21 is induced by the tumor suppressor p53. p27 responds to growth suppressors such as transforming growth factor-. |
| • 1NK4/ARF family: p16INK4A, p14ARF | p16INK4a binds to cyclin D-CDK4 and promotes the inhibitory effects of RB. p14ARF increases p53 levels by inhibiting MDM2 activity. |
| Checkpoint Components | |
| • p53 | Tumor suppressor altered in the majority of cancers; causes cell-cycle arrest and apoptosis. Acts mainly through p21 to cause cell-cycle arrest. Causes apoptosis by inducing the transcription of pro-apoptotic genes such as BAX. Levels of p53 are negatively regulated by MDM2 through a feedback loop. p53 is required for the G1 /S checkpoint and is a main component of the G2 /M checkpoint. |
| • Ataxia-telangiectasia mutated (ATM) | Activated by mechanisms that sense double stranded DNA breaks. Transmits signals to arrest the cell cycle after DNA damage. Acts through p53 in the G1 /S checkpoint. At the G2 /M checkpoint, it acts both through p53-dependent mechanisms and through the inactivation of CDC25 phosphatase, which disrupts the cyclin B-CDK1 complex. Component of a network of genes that include BRCA1 and BRCA2, which link DNA damage with cell-cycle arrest and apoptosis. |
Three components, p21, p27, and p57, which bind to and inactivate the complexes formed between cyclins and CDKs. Transcriptional activation of p21 is under the control of p53, a tumor suppressor gene that is mutated in a large proportion of human cancers. The main role of p53 in the cell cycle is one of surveillance, triggering checkpoint controls that slow down or stop cell-cycle progression of damaged cells, or causes apoptosis. The human INK4a/ARF locus (a notation for "inhibitor of kinase 4/alternative reading frame") encodes two proteins, p16INK4a and p14ARF, which block the cell cycle and act as tumor suppressors. p16INK4a competes with cyclin D for binding to CDK4 and inhibits the ability of the cyclin D-CDK4 complex to phosphorylate RB, thus causing cell-cycle arrest at late G1 . It is frequently mutated or inactivated by hypermethylation (discussed later) in human cancers. The INK4a locus encodes a second gene product, p14ARF (p19ARF in mice), which acts on p53. p14ARF arises from an alternative reading of the INK4a gene, providing for an "economical" way to utilize gene-coding sequences.[42] Although both p16INK4a and p14ARF block the cell cycle, their targets are different; p16INK4a acts on cyclin D-CDK4, whereas p14ARF prevents p53 degradation.
Date: 2016-04-22; view: 1180
| <== previous page | | | next page ==> |
| Defective DNA Repair Syndromes. | | | Protooncogenes, Oncogenes, and Oncoproteins |