CATEGORIES:
BiologyChemistryConstructionCultureEcologyEconomyElectronicsFinanceGeographyHistoryInformaticsLawMathematicsMechanicsMedicineOtherPedagogyPhilosophyPhysicsPolicyPsychologySociologySportTourism
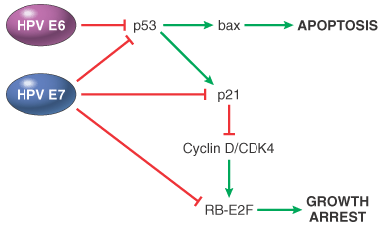
Human Papillomavirus.Approximately 70 genetically distinct types of HPV have been identified. Some types (e.g., 1, 2, 4, and 7) cause benign squamous papillomas (warts) in humans. Human papilloma viruses have been implicated in the genesis of several cancers, particularly squamous cell carcinoma of the cervix and anogenital region, and in some cases, to the causation of oral and laryngeal cancers ( Chapter 16 ).[164] Epidemiologic studies suggest that carcinoma of the cervix is caused by a sexually transmitted agent, and HPV is the culprit. DNA sequences of HPV 16 and 18 and, less commonly, HPV 31, 33, 35, and 51 are found in approximately 85% of invasive squamous cell cancers and their presumed precursors (severe dysplasias and carcinoma in situ). In contrast to cervical cancers, genital warts with low malignant potential are associated with distinct HPV types, predominantly HPV 6 and HPV 11 ("low-risk" types). Molecular analyses of HPV-associated carcinomas and benign genital warts reveal differences that may be pertinent to the transforming activity of these viruses. In benign warts and in preneoplastic lesions, the HPV genome is maintained in an episomal (nonintegrated) form, whereas in cancers, the viral DNA is usually integrated into the host cell genome. This suggests that integration of viral DNA is important in malignant transformation. Although the site of viral integration in host chromosomes is random (the viral DNA is found at different locations in different cancers), the pattern of integration is clonal; that is, the site of integration is identical within all cells of a given cancer. This would not occur if HPV were merely a passenger that infects cells after transformation. Furthermore, the viral DNA is interrupted at a fairly constant site in the process of integration: It is almost always within the E1/E2 open reading frame of the viral genome. Because the E2 region of the viral DNA normally represses the transcription of the E6 and E7 early viral genes, its interruption causes over-expression of the E6 and E7 proteins of HPV 16 and HPV 18. The oncogenic potential of HPV 16 and HPV 18 can be related to these two early viral gene products, which act in conjunction to immortalize and transform cells.[162] [165] The replication of DNA viruses is dependent on the replication machinery of the host cells, and E6 and E7 act to overcome the activity of cell-cycle inhibitors ( Fig. 7-50 ).[166] E6 binds to p53 and E7 binds to RB, inducing the degradation of these proteins. In addition, E7 can interfere with p53 transcriptional activity and also inactivate p21. Thus, E6 and E7 block p53 and RB cell cycle suppression pathways. The affinity of these viral proteins for the products of tumor suppressor genes differs depending on the oncogenic potential of HPV. E6 proteins derived from high-risk HPV (HPV 16, 18, and 31) inactivate p53 by enhancing its degradation through ubiquitin-dependent proteolysis.[167] E6 proteins of low-risk HPV (HPV 6 and 11) bind p53 with low affinity and have no effect on p53 stability. E7 proteins from high-risk HPV strongly bind to RB, disrupting the E2F/RB complex and promoting the degradation of RB. By contrast, E7 proteins from low-risk HPV have lower affinity for RB and have a weak capacity to transform cells. Thus, E6 and E7 proteins of high-risk HPV disable two important tumor suppressor proteins that regulate the cell cycle. In HPVinduced tumors, p53 mutations are extremely uncommon, presumably because loss of p53 function is accomplished by binding to the E6 oncoprotein. This binding not only blocks the inhibitory effect of p53 on the cell cycle but also interferes with p53 activation after DNA damage, a mechanism that allows DNA repair or elimination of cells with genomic damage. Moreover, E6 may have other effects independent of its binding of p53, such as the activation of telomerase and tyrosine kinases.[164]
Figure 7-50Effect of HPV proteins E6 and E7 on the cell cycle. E6 and E7 enhance p53 degradation, causing a block in apoptosis and decreased activity of the p21 cell cycle inhibitor. E7 associates with p21 and prevents its inhibition of the Cyclin/CDK4 complex; E7 can bind to RB, removing cell cycle restriction. The net effect of HPV E6 and E7 proteins is to block apoptosis and remove the restrains to cell proliferation (see Fig. 7-29 ). (Modified from Münger K, Howley PM: Human papillomavirus immortalization and transformation functions. Virus Research 89:213–228, 2002.)
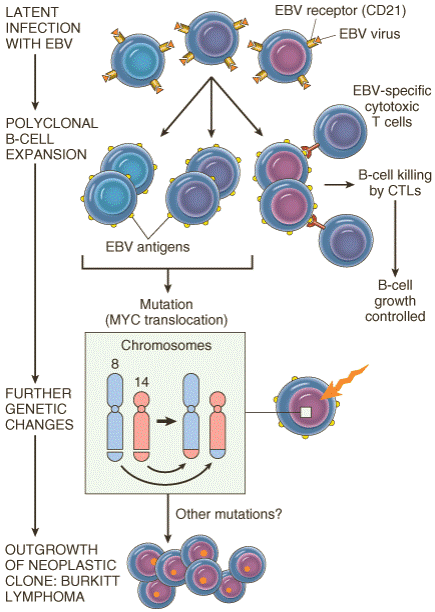
Figure 7-51Schema depicting the possible evolution of Epstein-Barr virus (EBV)-induced Burkitt lymphoma.
Figure 7-52Tumor antigens recognized by CD8+ T cells. (Modified from Abbas AK, Lichtman AH: Cellular and Molecular Immunology, 5th ed. Philadelphia, WB Saunders, 2003.)
Figure 7-53Mechanisms by which tumors evade the immune system. (Reprinted from Abbas AK, Lichtman AH: Cellular and Molecular Immunology, 5th ed. Philadelphia, WB Saunders, 2003.)
TABLE 7-12-- Paraneoplastic Syndromes Date: 2016-04-22; view: 714
|