CATEGORIES:
BiologyChemistryConstructionCultureEcologyEconomyElectronicsFinanceGeographyHistoryInformaticsLawMathematicsMechanicsMedicineOtherPedagogyPhilosophyPhysicsPolicyPsychologySociologySportTourism
Box 7-1. Gene Expression Profiles of Human Cancers. Microarrays and Proteomics
Until recently, studies of gene expression in tumors involved the analysis of individual genes. These studies have been revolutionized by the introduction of methods that can measure the expression of thousands of genes simultaneously. [208] [209] The most common method for large-scale analysis of gene expression in use today is based on DNA microarray technology. In this method, DNA fragments, either cDNAs or oligonucleotides, are spotted on a glass slide or on some other solid support. As the techniques used for the spotting are similar to those employed to produce semiconductor chips for electronic products, the arrays are known as "gene chips." Chips can be purchased from commercial suppliers or produced in-house, and can contain more than 20,000 gene fragments. The fragments are typically obtained from complementary DNA (cDNA) libraries or sets of nucleotides from known and uncharacterized genes. The gene chip is then hybridized to "probes" prepared from tumor and control samples (the probes are usually cDNA copies of RNAs extracted from tumor and uninvolved tissues). Before hybridization to the chip, the probes are labeled with fluorochromes that emit different colors (e.g. red color for tumor RNA and green color for control RNA). After hybridization the chip is read using a laser scanner ( Fig. 7-45 ); each spot on the array will be red (increased expression of a gene in the tumor), green (decreased expression in the tumor) or, if there is no difference in gene expression between the tumor and control sample, the spots will be either black or yellow (depending on the type of fluorescent scanning). Sophisticated software has been developed to measure the intensity of the fluorescence for each spot and produce data sets in which genes with similar expression patterns are clustered. [210] This method of analysis, called hierarchical clustering, groups together genes according to the similarity of their gene expression patterns. The software can be linked to large sequencing and array databases available through the Web. This allows appropriate gene identification and comparison between expression profiles from various sources. A major problem in the analysis of gene expression in tumors is the heterogeneity of the tissue. In addition to the heterogeneity between tumor cells, samples may contain variable amounts of stromal connective tissue, inflammatory infiltrates, and normal tissue cells. One way to overcome this problem is to obtain nearly pure tumor cells or small tumors free from associated tissues using laser capture microdissection. In this technique, the dissection of the tumor or cells is made under a microscope through a focused laser. The dissected material is then captured or "catapulted" into a small cap and processed for RNA and DNA isolation.
Gene expression profiling of tumors has multiple uses, and the number of publications using this technique has grown enormously during the past few years. Much of the work performed is not directed toward proving or disproving a proposed hypothesis. Gene expression analysis can be used to classify tumors; to predict metastatic potential, prognosis, and response to therapy; to reveal gene expression patterns that are dependent on the mutation of a single oncogene; and to analyze the effects of hormones and environmental agents on cancer development. [209] The applications of this technology keep expanding and being refined, but much has already been accomplished.[141] [209] [210] [211] We mention only a few interesting examples. Profiling of cells from adult and pediatric T-cell acute lymphoblastic leukemia has identified the patterns of gene expression in leukemic blast cells and has accurately classified each prognostic subtype.[211] The work that has received the highest publicity involves gene expression profiling of breast cancers. In addition to identifying new subtypes of breast cancers, a 70-gene prognosis profile was established. Using this type of profile, it has been reported that: (1) the profile was a powerful predictor of disease prognosis for young patients; (2) it was particularly accurate for predicting metastasis during the first 5 years after diagnosis; and (3) prognosis determined by gene expression profiles correlated highly with histologic grade and estrogen receptor status but not with lymphatic spread of the tumor.[211] A more recent analysis has pooled together data gathered by different laboratories and has confirmed the identification of distinct subtypes of breast cancer.[142] Given all of these remarkable results, it is time to ask whether this technology is "ready for prime time"; that is, ready for day to day clinical applications. Things are moving very fast in this area, but before clinical applications are considered, many issues need to settled. Not only do larger trials need to be conducted to prove the reliability and accuracy of the analysis but also, just as important, the procedures for handling samples, performing the analyses, and reporting the data need to be standardized, so that data obtained in various laboratories can be compared.
Next on the horizon of molecular techniques for the global analysis of gene expression in cancers is proteomics, a technique used to obtain expression profiles of proteins contained in tissues, serum, or other body fluids. The original method consisted of the separation of proteins by 2-dimensional gel electrophoresis, followed by identification of individual proteins by mass spectrometry. A more recent technique, called ICAT (isotope-coding affinity tags) does not rely on electrophoresis for protein separation. In ICAT, proteins in the test and control samples are labeled with light or heavy isotopes. The differentially labeled proteins are then identified and quantified by mass spectrometry. A variation of proteomic analysis has been used to obtain protein profiles in the blood of cancer patients without identification of individual proteins.[215]
The excitement created by the development of new techniques for the global molecular analysis of tumors has led some scientists to predict that the end of histopathology is in sight, and to consider existing approaches to tumor diagnosis as the equivalent of magical methods of divination. Indeed, it is hard to escape the excitement generated by the development of entirely new and powerful methods of molecular analysis. However, what lies ahead is not the replacement of one set of techniques by another. On the contrary, the most accurate diagnosis and prognosis of cancer will be arrived at by a combination of morphologic and molecular techniques. [208]

Figure 7-45Schematic representation of the steps required for the analysis of global gene expression by DNA microarray. RNA is extracted from tumor and normal tissue. cDNA synthesized from each preparation is labeled with fluorescent dyes (in the example shown, normal tissue cDNA is labeled with a green dye; tumor cDNA is labeled with a red dye). The array consists of a solid support in which DNA fragments from many thousands of genes are spotted. The labeled cDNAs from tumor and normal tissue are combined and hybridized to the genes contained in the array. Hybridization signals are detected using a confocal laser scanner and downloaded to a computer for analysis (red squares, expression of the gene is higher in tumor; green square, expression of the gene is higher in normal tissue; black squares, no difference in the expression of the gene between tumor and normal tissue). In the display, the horizontal rows correspond to each gene contained in the array; each ventrical row corresponds to single samples.

Figure 7-47Schematic illustration of the pathways of malignancy initated by mutation of the gatekeeper genes (e.g., APC, NF-1, RB) or caretaker genes (e.g., hMSH2, BRCA-1, BRCA-2).

Figure 7-48Experiments demonstrating the initiation and promotion phases of carcinogenesis in mice. Group 2: application of promoter repeated at twice-weekly intervals for several months. Group 3: application of promoter delayed for several months and then applied twice weekly. Group 6: promoter applied at monthly intervals.
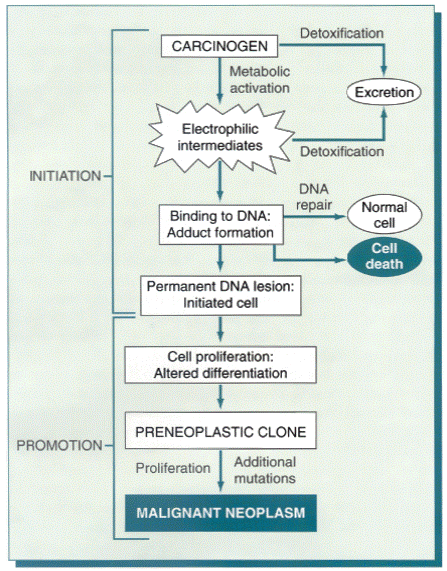
Figure 7-49General schema of events in chemical carcinogenesis. Note that promoters cause clonal expansion of the initiated cell, thus producing a preneoplastic clone. Further proliferation induced by the promoter or other factors causes accumulation of additional mutations and emergence of a malignant tumor.
TABLE 7-11-- Major Chemical Carcinogens
Date: 2016-04-22; view: 938
| <== previous page | | | next page ==> |
| Malignancy Translocation Affected Genes | | | Direct-Acting Carcinogens |