CATEGORIES:
BiologyChemistryConstructionCultureEcologyEconomyElectronicsFinanceGeographyHistoryInformaticsLawMathematicsMechanicsMedicineOtherPedagogyPhilosophyPhysicsPolicyPsychologySociologySportTourism
Subcellular Location Gene Function
Tumors Associated with Somatic
Mutations
Tumors Associated with Inherited
Mutations
Cell surface TGF-receptor Growth inhibition Carcinomas of colon Unknown
E-cadherin Cell adhesion Carcinoma of stomach Familial gastric cancer
Inner aspect of plasma membrane NF-1 Inhibition of RAS signal
transduction and of p21 cell-cycle
inhibitor
Neuroblastomas Neurofibromatosis type 1 and sarcomas
Cytoskeleton NF-2 Cytoskeletal stability Schwannomas and meningiomas Neurofibromatosis type 2, acoustic
schwannomas and meningiomas
Cytosol APC/-catenin Inhibition of signal transduction Carcinomas of stomach, colon,
pancreas; melanoma
Familial adenomatous polyposis coli/
colon cancer
PTEN PI-3 kinase signal transduction Endometrial and prostate cancers Unknown
SMAD 2 and SMAD 4 TGF-signal transduction Colon, pancreas tumors Unknown
Nucleus RB Regulation of cell cycle Retinoblastoma; osteosarcoma
carcinomas of breast, colon, lung
Retinoblastomas, osteosarcoma
p53 Cell-cycle arrest and apoptosis in
response to DNA damage
Most human cancers Li-Fraumeni syndrome; multiple
carcinomas and sarcomas
WT-1 Nuclear transcription Wilms tumor Wilms tumor
p16 (INK4a) Regulation of cell cycle by
inhibition of cyclin-dependent
kinases
Pancreatic, breast, and esophageal
cancers
Malignant melanoma
BRCA-1 and BRCA-2 DNA repair Unknown Carcinomas of female breast and ovary;
carcinomas of male breast
KLF6 Transcription factor Prostate Unknown
Because RAS is so frequently mutated in human cancers, much effort has been spent to develop anti-RAS modalities of targeted therapy. Several such strategies for cancer treatment are being evaluated. The specific targets include blockade of ( Fig. 7-36 ). When cells enter the S phase, they can continue to cell division independent of growth factors. It should be obvious from this discussion that if RB is absent (owing to gene deletions) or its ability to regulate E2F transcription factors is derailed, the molecular brakes on the cell cycle are released, and the cells move into the S phase followed by cell replication.
The mutations of RB genes found in tumors are localized to a region of the RB protein, called the "RB pocket," that is involved in binding to E2F.
It was mentioned previously that germ-line loss or mutations of the RB gene predispose to occurrence of retinoblastomas and to a lesser extent osteosarcomas. Furthermore, somatically acquired mutations have been described in glioblastomas, small cell carcinomas of lung, breast cancers, and bladder carcinomas. Given the presence of RB in every cell and its importance in cell-cycle control, two questions arise: (1) Why do patients with germ line mutation of the RB locus develop mainly retinoblastomas? (2) Why are inactivating mutations of RB not much more common in human cancer? The basis for the occurrence of tumors restricted to the retina in patients who inherit one defective allele of RB is not fully understood, but some possible explanations have emerged from the study of mice with targeted disruption of the RB locus. For instance, RB mutation may be a critical initiating event for retinoblastomas but may be only an accessory factor for malignancies at other sites.
With respect to the second question (i.e., why the loss of RB is not more common in human tumors), the answer is much simpler: Mutations in other genes that control RB phosphorylation can mimic the effect of RB loss, and such genes are mutated in many cancers that may have normal RB genes. Thus, for example, mutational activation of cyclin D or CDK4 would favour cell proliferation by facilitating RB phosphorylation. As previously discussed, cyclin D is overexpressed in many tumors because of gene amplification or translocation. Mutational inactivation of CDK inhibitors would also drive the cell cycle by unregulated activation of cyclins and CDKs. Thus, the emerging paradigm is that loss of normal cell-cycle control is central to malignant transformation and that at least one of four key regulators of the cell cycle (p16INK4a, CYCLIN D, CDK4, RB) is dysregulated in the vast majority of human cancers. [38] In cells that harbor mutations in any one of these other genes, the function of RB is disrupted even if the RB gene itself is not mutated.[45]
Several other pathways of cell growth regulation, some to be discussed in more detail later, also converge on RB ( Fig. 7-36 ):
• TGF-induces inhibition of cellular proliferation. This effect of TGF-is mediated, at least in part, by up-regulation of the CDK inhibitor p27.
• The transforming proteins of several oncogenic animal and human DNA viruses seem to act, in part, by neutralizing the growth inhibitory activities of RB. In these cases, RB protein is functionally deleted by the binding of a viral protein and no longer acts as a cell-cycle inhibitor. Simian virus 40 and polyomavirus large T antigens, adenoviruses EIA protein, and human papillomavirus (HPV) E7 protein, all bind to the hypophosphorylated form of RB. The binding occurs in the same RB pocket that normally sequesters E2F transcription factors; in the case of HPV, the binding is particularly strong for viral types, such as HPV 16, which confer high risk for the development of cervical carcinomas. Thus, the RB protein, unable to bind the E2F transcription factors, is functionally deleted, and the transcription factors are free to cause cell-cycle progression.
• The p53 tumor suppressor gene exerts its growth-inhibiting effects at least in part by up-regulating the synthesis of the CDK inhibitor p21 (see Fig. 7-29 and Fig. 7-36 ).

Figure 7-35Pathogenesis of retinoblastoma. Two mutations of the RB locus on chromosome 13q14 lead to neoplastic proliferation of the retinal cells. In the familial form, all somatic cells inherit one mutant RB gene from a carrier parent. The second mutation affects the Rb locus in one of the retinal cells after birth. In the sporadic form, on the other hand, both mutations at the RB locus are acquired by the retinal cells after birth.

Figure 7-36Role of RB as a cell-cycle regulator. Various growth factors promote the formation of the cyclin D-CDK4 complex. This complex (and to some extent cyclin E-CDK2) phosphorylates RB, changing it from an active (hypophosphorylated) to an inactive state (hyperphosphorylation). RB inactivation allows the cell to pass the G1 /S restriction point. Growth inhibitors such as TGF-and p53 and the Cip/Kip (e.g., p21, p57) and INK4a (p161NK4a and p19ARF) cell-cycle inhibitors prevent RB activation. Transforming proteins of oncogenic viruses bind hypophosphorylated RB and cause its functional inactivation. Virtually all cancers show dysregulation of the cell cycle by affecting the four genes marked by an asterisk.

Figure 7-37The role of p53 in maintaining the integrity of the genome. Activation of normal p53 by DNA-damaging agents or by hypoxia leads to cell-cycle arrest in G1 and induction of DNA repair, by transcriptional up-regulation of the cyclin-dependent kinase inhibitor p21, and the GADD45 genes, respectively. Successful repair of DNA allows cells to proceed with the cell cycle; if DNA repair fails, p53-induced activation of the BAX gene promotes apoptosis. In cells with loss or mutations of p53, DNA damage does not induce cell-cycle arrest or DNA repair, and hence genetically damaged cells proliferate, giving rise eventually to malignant neoplasms.

Figure 7-38 A, The role of APC in regulating the stability and function of -catenin. APC and -catenin are components of the WNT signaling pathway. In resting cells (not exposed to WNT), -catenin forms a macromolecular complex containing the APC protein. This complex leads to the destruction of -catenin, and intracellular levels of -catenin are low. B, When cells are stimulated by secreted WNT molecules, the destruction complex is deactivated, -catenin degradation does not occur, and cytoplasmic levels increase. -catenin translocates to the nucleus, where it binds to TCF, a transcription factor that activates several genes involved in the cell cycle. C, When APC is mutated or absent, the destruction of -catenin cannot occur. - Catenin translocates to the nucleus and coactivates genes that promote the cell cycle, and cells behave as if they are under constant stimulation by the WNT pathway.
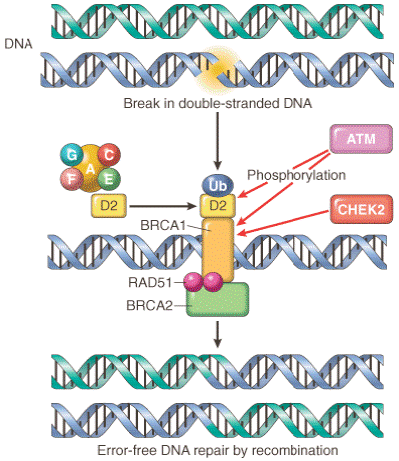
Figure 7-39Interaction between cancer susceptibility genes and DNA repair. ATM (ataxia-telangiectasia mutated) senses a double-strand break in DNA, induced by agents such as ionizing
radiation. ATM and CHEK2 phosphorylate BRCA1, promoting its migration to the break site. The Fanconi's anemia protein complex (proteins A, C, E, F, G) triggers the ubiquitination and colocalization of the Fanconi protein D2 with BRCA1 at the break site. BRCA2 carries RAD51, an enzyme involved in DNA recombination repair, to the same site. BRCA1, BRCA2, and RAD51 repair the DNA break by an error-free recombination mechanism. RAD51 is a component of cell cycle check points. (Redrawn from Venkitaraman AR: A growing network of cancer-susceptibility genes. N Engl J Med 348:1917, 2003.)

Figure 7-40Cellular responses to telomere shortening. The figures show the responses of normal cells, which have intact cell-cycle checkpoints and of cells with checkpoint defects.
(From Wong JMY, Collins K: Telomere maintenance and disease. Lancet 362:983, 2003.)
Figure 7-42The metastatic cascade. Schematic illustration of the sequential steps involved in the hematogenous spread of a tumor.

Figure 7-43Mechanisms of metastasis development within a primary tumor. A nonmetastatic primary tumor is shown (light blue) on the left side of all diagrams. Four models are presented: A, Metastasis is caused by rare variant clones that develop in the primary tumor; B, Metastasis is caused by the gene expression pattern of most cells of the primary tumor, referred to as a metastatic signature; C, A combination of A and B, in which metastatic variants appear in a tumor with a metastatic gene signature; D, Metastasis development is greatly influenced by the tumor stroma, which may regulate angiogenesis, local invasiveness and resistance to immune elimination, allowing cells of the primary tumor, as in C, to become metastatic.

Figure 7-44A–D, Schematic illustration of the sequence of events in the invasion of epithelial basement membranes by tumor cells. Tumor cells detach from each other because of reduced adhesiveness, and cells then attach to the basement membrane via the laminin receptors and secrete proteolytic enzymes, including type IV collagenase and plasminogen activator. Degradation of the basement membrane and tumor cell migration follow.
TABLE 7-10-- Selected Examples of Oncogenes Activated by Translocation
Date: 2016-04-22; view: 833
| <== previous page | | | next page ==> |
| Signal-Transducing Proteins. | | | Malignancy Translocation Affected Genes |